
The race to accelerate drug discovery just got a significant boost. Optibrium’s release of a user-friendly PyMOL plugin for its QuanSA binding affinity prediction method isn’t just a software update; it’s a signal of a broader shift towards AI-driven, physics-informed approaches that are democratizing access to advanced computational chemistry. For years, accurate binding affinity prediction was the domain of large pharma with massive compute resources. Now, smaller biotechs and academic labs can leverage similar predictive power, potentially slashing development timelines and costs.
- Democratization of Prediction: QuanSA, previously a command-line tool, is now accessible to a wider range of chemists via a visual interface.
- Cost & Time Savings: The method offers accuracy comparable to computationally expensive techniques like FEP, but at a fraction of the cost and without requiring a protein structure.
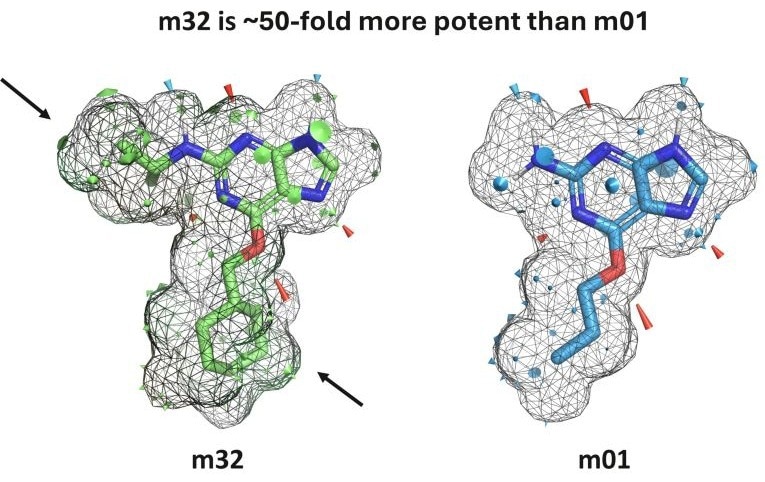
- Insight Beyond Strength: The plugin visualizes *why* a molecule binds, not just *how strongly*, enabling more informed design decisions.
Traditionally, predicting how well a drug candidate will bind to its target has been a bottleneck in the early stages of drug discovery. Methods like free energy perturbation (FEP) are highly accurate, but demand significant computational power and expertise. Other methods often rely on having a detailed protein structure, which isn’t always available. Optibrium’s QuanSA tackles both these challenges. It uses a physically-motivated machine learning approach – meaning it’s grounded in the fundamental principles of chemistry and physics, rather than being a purely ‘black box’ AI – and crucially, it can function even without a complete protein structure. This is a game-changer for targeting proteins where structural data is limited, a common scenario in emerging therapeutic areas.
The move to integrate QuanSA with PyMOL, a widely used molecular visualization tool, is particularly astute. It lowers the barrier to entry for chemists who may not be computational experts. Optibrium’s previous release of a PyMOL interface for Surflex-Dock demonstrates a clear strategy: to embed sophisticated modeling capabilities directly into the workflows of medicinal chemists. This isn’t about replacing experts; it’s about augmenting their abilities and empowering a broader range of scientists to leverage these tools.
The Forward Look
This release isn’t an isolated event. Expect to see increased pressure on software vendors to provide similar “AI-powered accessibility” for complex modeling tasks. The success of QuanSA will likely spur competitors to develop comparable solutions, potentially leading to a rapid evolution of the computational chemistry landscape. More importantly, the focus will shift from simply *predicting* binding affinity to *understanding* the underlying biophysics. We’ll likely see further integration of these predictive tools with automated synthesis platforms, creating closed-loop design-build-test cycles that dramatically accelerate lead optimization. The real question isn’t whether AI will transform drug discovery – it already is – but how quickly these advancements will translate into novel therapies reaching patients. The next step for Optibrium, and the industry, will be demonstrating how these early predictions translate into a higher success rate in clinical trials. That’s the ultimate metric.
Discover more from Archyworldys
Subscribe to get the latest posts sent to your email.