
A newly identified genetic cause of neurodevelopmental disorders (NDDs) – stemming from mutations in the RNU2-2 gene – is poised to reshape diagnostic pathways and potentially unlock novel therapeutic strategies. Researchers have, for the first time, definitively linked recessive mutations in RNU2-2 to a distinct NDD syndrome, demonstrating a surprisingly high prevalence within the 100,000 Genomes Project and subsequent validation studies. This isn’t simply another gene added to the list of NDD causes; the unique mechanism of action and the potential for targeted interventions set it apart.
Key Takeaways
- High Prevalence: Recessive RNU2-2 syndrome is a surprisingly common genetic cause of NDDs, accounting for a significant proportion of cases in the UK’s genomic datasets – potentially up to 13.1% of all NDDs with a biallelic genetic diagnosis.
- Unique Mechanism: The disorder appears to stem from impaired U2 snRNA function, leading to reduced U2-2 expression and potentially disrupting the splicing process, but without widespread aberrant splicing detected thus far.
- Diagnostic Implications: The identification of ‘tier 1’ and ‘tier 2’ variants, coupled with U2-2 expression analysis, offers a new diagnostic avenue for individuals with unexplained NDDs, particularly those with features like seizures, developmental delay, and microcephaly.
The Deep Dive: Unraveling the RNU2-2 Connection
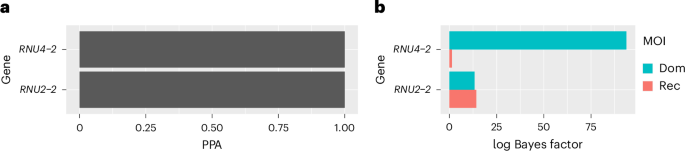
For years, the landscape of NDD genetics has been a frustratingly complex puzzle. While many genes have been implicated, a substantial proportion of cases remain undiagnosed. This research, leveraging the power of large-scale genomic data from the UK’s 100,000 Genomes Project and international collaborations, provides a crucial piece. The team employed a sophisticated genetic association method, BeviMed, to pinpoint RNU2-2 as a key player. Crucially, they distinguished between dominant and recessive inheritance patterns, revealing that recessive mutations are the primary driver of this specific NDD.
RNU2-2 encodes a small non-coding RNA molecule vital for mRNA splicing – a fundamental process in gene expression. The identified mutations largely disrupt the internal structure of the U2 snRNA, potentially hindering its ability to interact with other splicing components. Interestingly, individuals with biallelic RNU2-2 mutations exhibit significantly reduced levels of U2-2 RNA, and a compensatory increase in U2-1 RNA, suggesting a cellular attempt to maintain overall U2 snRNA levels. This finding is a critical biomarker, offering a potential avenue for diagnostic testing.
The clinical presentation of recessive RNU2-2 syndrome is varied, ranging from mild learning disabilities to severe epileptic encephalopathy. Common features include hypotonia, global developmental delay, and seizures. While a distinct facial gestalt hasn’t been identified, some individuals exhibit subtle dysmorphic features. The researchers meticulously categorized cases into ‘tier 1’ (high confidence) and ‘tier 2’ (lower confidence) based on variant pathogenicity scores and clinical data, providing a framework for clinical interpretation.
The Forward Look: What Happens Next?
The identification of RNU2-2 as a significant cause of NDDs opens several exciting avenues for future research and clinical practice. First, we can expect a rapid expansion of diagnostic testing. The availability of a clear genetic target and a measurable biomarker (U2-2 expression) will enable more accurate and timely diagnoses for affected individuals. Laboratories will likely incorporate RNU2-2 sequencing and RNA expression analysis into their NDD diagnostic panels.
Second, the unique mechanism of action – disruption of the spliceosome – presents a potential therapeutic target. While directly restoring U2 snRNA function is a significant challenge, researchers may explore strategies to modulate splicing pathways or compensate for the loss of U2-2 function. The discovery of the compensatory upregulation of U2-1 is particularly intriguing and warrants further investigation. Could manipulating U2-1 expression offer a therapeutic benefit?
Third, the observed prevalence of de novo mutations in RNU2-2 suggests a relatively high mutation rate, potentially linked to the polymerase III machinery involved in snRNA transcription. Further investigation into the mutational landscape of RNU2-2 and related genes could reveal insights into the underlying mechanisms driving these mutations.
Finally, the study highlights the power of large-scale genomic initiatives like the 100,000 Genomes Project. These projects are not just about identifying genes; they are about building a foundation for precision medicine and transforming the lives of individuals affected by rare diseases. Expect to see increased investment in similar initiatives globally, accelerating the pace of discovery in the field of NDD genetics.
Discover more from Archyworldys
Subscribe to get the latest posts sent to your email.